 Việt Nam | Life Science & GMP Solutions") SAO NAM | Particle Measuring Systems (PMS) Việt Nam | Life Science & GMP Solutions
SAO NAM | Particle Measuring Systems (PMS) Việt Nam | Life Science & GMP Solutions






_-02-01-2024-11-57-38.jpg "Thiết kế phòng sạch dược phẩm cơ bản")

 Việt Nam | Life Science & GMP Solutions")

Toàn vẹn dữ liệu & 21 CFR Part 11 - Hiểu và Tuân thủ các yêu cầu của GMP và FDA
-
Hãng sản xuất: Particle Measuring Systems - Mỹ Model: Document: -
Liên hệ

Toàn vẹn dữ liệu và 21 CFR Part 11
21 CFR Part 11: Toàn vẹn dữ liệu, hồ sơ điện tử và chữ ký điện tử trong ngành dược phẩm
21 CFR Part 11 là một phần trong Bộ Quy định Liên bang Hoa Kỳ do FDA ban hành, quy định cách quản lý hồ sơ điện tử và chữ ký điện tử để chúng có thể được chấp nhận như hồ sơ giấy và chữ ký viết tay trong các hoạt động được quản lý.
Với ngành dược phẩm, 21 CFR Part 11 thường được nhắc đến trong bối cảnh toàn vẹn dữ liệu, hệ thống máy tính, phần mềm giám sát, dữ liệu phòng sạch, dữ liệu kiểm nghiệm, dữ liệu sản xuất, dữ liệu hiệu chuẩn và các hệ thống điện tử có ảnh hưởng đến chất lượng sản phẩm.
Trong nhà máy dược phẩm, dữ liệu là bằng chứng chứng minh sản phẩm được sản xuất, kiểm tra, bảo quản và giám sát trong điều kiện được kiểm soát. Nếu dữ liệu không đáng tin cậy, bị chỉnh sửa không kiểm soát, thiếu người chịu trách nhiệm hoặc không truy xuất được lịch sử, doanh nghiệp sẽ gặp rủi ro lớn khi audit, điều tra sai lệch hoặc đánh giá chất lượng sản phẩm.
Đối với phòng sạch, dữ liệu từ máy đếm tiểu phân, hệ thống giám sát online, máy lấy mẫu vi sinh, dữ liệu APSS-2000, báo cáo DataAnalyst hoặc FMS đều có thể trở thành dữ liệu quan trọng trong hồ sơ GMP. Vì vậy, dữ liệu cần rõ ràng, đầy đủ, bảo mật, có audit trail, được sao lưu và được quản lý theo SOP phù hợp.
Trong phòng sạch, máy đếm tiểu phân và hệ thống giám sát môi trường tạo ra dữ liệu phục vụ kiểm soát nhiễm bẩn. Dữ liệu này có thể được dùng để đánh giá cấp độ sạch, giám sát xu hướng, điều tra cảnh báo, chứng minh điều kiện sản xuất hoặc hỗ trợ hồ sơ audit. Vì vậy, cách dữ liệu được tạo, lưu, sửa, xuất và phê duyệt cần được kiểm soát chặt chẽ.
Các thiết bị và phần mềm hiện đại như Lasair Pro, DataAnalyst, FMS, Pharmaceutical Net Pro, Airnet, IsoAir, MiniCapt hoặc APSS-2000 thường được lựa chọn không chỉ vì khả năng đo, mà còn vì khả năng hỗ trợ quản lý dữ liệu, người dùng, recipe, location, báo cáo, audit trail và toàn vẹn dữ liệu.
21 CFR Part 11 defines FDA requirements for electronic records and electronic signatures. In pharmaceutical cleanroom monitoring, it is closely related to data integrity, audit trail, user access control, secure data storage, electronic reports and traceable environmental monitoring records.
SAO NAM supports customers in Vietnam with Particle Measuring Systems instruments and software, including Lasair Pro, Airnet, IsoAir, MiniCapt, APSS-2000, DataAnalyst, FMS and related calibration, maintenance and technical services.
For PMS equipment consultation, data integrity support, cleanroom monitoring solutions, calibration, repair or technical assistance, please contact SAO NAM.
Dòng máy đếm tiểu phân Lasair Pro
Lasair Pro 310 - Máy đếm tiểu phân 28.3 LPM
IsoAir Pro Plus - Máy đếm tiểu phân online
Airnet 510 - Sensor đếm tiểu phân online
MiniCapt Remote - Máy lấy mẫu vi sinh online
APSS-2000 - Máy đếm tiểu phân trong dung dịch
Chức năng Audit Trail trong hệ thống dữ liệu
Hiệu chuẩn thiết bị Particle Measuring Systems
Hiểu và Tuân thủ các yêu cầu của GMP và FDA trong ngành Dược Phẩm
Data Integrity / Tính toàn vẹn dữ liệu đồng nghĩa với việc dữ liệu phải chính xác, đầy đủ và có thể lặp lại, điều này đảm bảo chất lượng sản phẩm và an toàn cho công chúng. Trong những năm gần đây, các vi phạm liên quan đến tính toàn vẹn dữ liệu đã được ghi nhận trong một số lá thư cảnh báo của Cục Quản lý Thực phẩm và Dược phẩm Hoa Kỳ (FDA), nhưng đây không phải là một khái niệm mới. Tầm quan trọng của việc ghi chép trong sản xuất dược phẩm có thể được truy tìm từ xa xưa, từ năm 1938, khi Đạo luật Thực phẩm, Dược phẩm và Mỹ phẩm Liên bang (FDC) yêu cầu phải ghi chép lại sự an toàn của các loại thuốc mới trước khi chúng được bán ra công chúng, với các quy định tương tự được áp dụng ở Châu Âu và Nhật Bản trong suốt thế kỷ 20.
Các hệ thống sản xuất có những rủi ro vận hành lớn và khó kiểm chứng. Thay vì chỉ phản ứng với các thảm họa sức khỏe công cộng, các biện pháp phòng ngừa, như yêu cầu chứng minh các tuyên bố, được thực hiện nhằm giảm thiểu khả năng xảy ra của chúng và tăng cường lòng tin cho nhà sản xuất
Sự Hiện Diện trong Các Quy Định

Trong vài năm qua, đã có nhiều lá thư cảnh báo của FDA (483s) được phát hành do thiếu sót về tính toàn vẹn dữ liệu trong ngành công nghiệp dược phẩm. Vào năm 2016, hơn 50% lá thư cảnh báo của MHRA liên quan đến sự cố về tính toàn vẹn dữ liệu đối với các hệ thống máy tính so với năm trước đó. Các thanh tra viên được đào tạo tích cực về các yêu cầu tính toàn vẹn dữ liệu và thực thi chúng một cách mạnh mẽ đối với các hồ sơ lô hàng bị giả mạo hoặc việc loại bỏ dữ liệu thô.
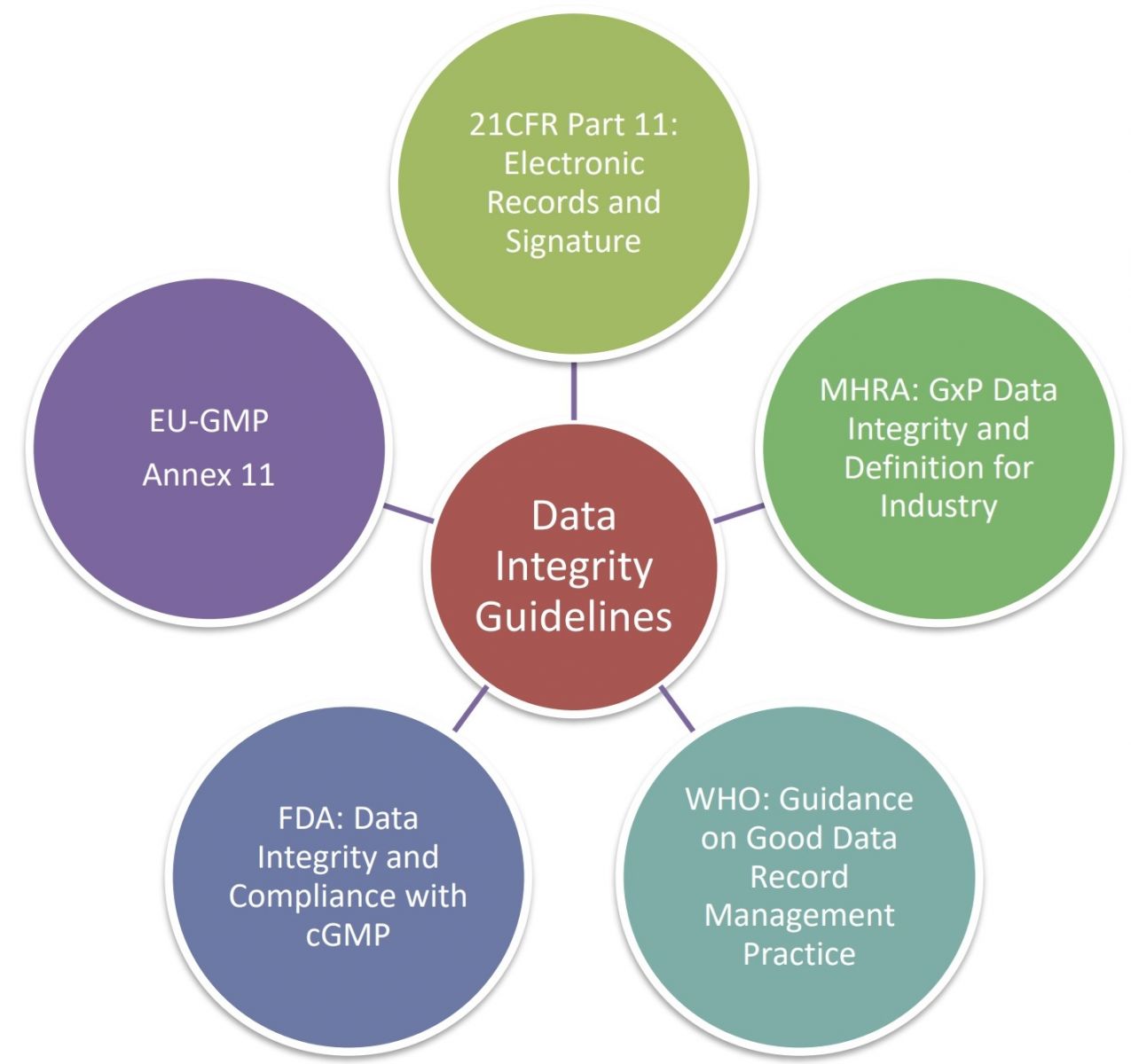
Việc hiểu biết về các tiêu chuẩn, hướng dẫn và quy định gần đây liên quan đến tính toàn vẹn dữ liệu là cần thiết để trở nên tuân thủ.
Các tài liệu liên quan bao gồm 21 CFR Phần 11, MHRA: GxP, EU GMP Phụ lục 1, FDA về Tính Toàn Vẹn Dữ Liệu và Tuân Thủ cGMP, và WHO về Thực Hành Quản Lý Dữ Liệu và Hồ Sơ Tốt. Mục tiêu cốt lõi của việc tuân thủ tính toàn vẹn dữ liệu là tăng cường chất lượng sản phẩm, lòng tin của cơ quan quản lý, uy tín thương hiệu và kiểm soát quy trình trong khi giảm thiểu khuyết tật sản phẩm và chi phí. Điều này áp dụng cho nhiều lĩnh vực trong ngành công nghiệp dược phẩm, bao gồm các nhà sản xuất sản phẩm dược phẩm hoàn chỉnh cho các thử nghiệm lâm sàng, nghiên cứu tương đương sinh học và phân phối thương mại, phòng thí nghiệm, sản xuất hợp đồng, nhà cung cấp, v.v.

1 -Tìm hiểu 21 CFR Part 11? 1. Nó là gì?
-
* Nó là gì? 21 CFR Part 11 là một phần của Tiêu đề 21 trong Luật Quy định Liên bang của Cơ quan Quản lý Thực phẩm và Dược phẩm Hoa Kỳ (FDA). Đây là tiêu chuẩn phổ biến nhất được sử dụng để quản lý dữ liệu một cách thích hợp. Part 11 áp dụng cho các hồ sơ được tạo, sửa đổi, duy trì, lưu trữ, truy xuất hoặc truyền thông theo yêu cầu được quy định bởi FDA trong định luật. Các hồ sơ điện tử/chữ ký điện tử đáp ứng các yêu cầu của Part 11 có thể được sử dụng thay cho hồ sơ giấy.
-
** Tại sao cần nó? 21 CFR Part 11 cần thiết để đảm bảo tính toàn vẹn và tuân thủ của dữ liệu điện tử trong các ngành liên quan đến sản xuất, lưu trữ và quản lý sản phẩm dược phẩm. Nó giúp đảm bảo rằng dữ liệu điện tử được tạo ra và sử dụng tuân thủ các quy định và yêu cầu của FDA, đặc biệt là trong việc đảm bảo chất lượng sản phẩm và an toàn của người tiêu dùng.
-
***Dùng nó như thế nào? Các tổ chức trong ngành sản xuất dược phẩm cần phải tuân thủ các quy định và yêu cầu của 21 CFR Part 11 khi sử dụng hồ sơ và chữ ký điện tử. Họ cần phải thực hiện các biện pháp bảo mật và kiểm tra để đảm bảo tính toàn vẹn và đáng tin cậy của dữ liệu điện tử. Nếu các hồ sơ và chữ ký điện tử đáp ứng các yêu cầu của Part 11, chúng có thể được sử dụng thay thế cho hồ sơ giấy.
21 CFR 11 Challenge: From Paper to Paperless - Webinars
2. Tại sao quan trọng trong ngành dược phẩm?
Quy định này quan trọng trong ngành dược phẩm vì:
-
Đảm bảo Tính Toàn Vẹn Dữ Liệu: Tránh tình trạng làm giả, thay đổi hoặc mất mát dữ liệu, đặc biệt là dữ liệu liên quan đến phát triển, sản xuất và kiểm tra sản phẩm dược.
- Tuân Thủ Pháp Lý: Giúp các doanh nghiệp tuân thủ các quy định của FDA, tránh rủi ro pháp lý và hậu quả nghiêm trọng như thu hồi sản phẩm hoặc phạt tiền.
- Chất Lượng và An Toàn Sản Phẩm: Bảo đảm chất lượng và an toàn của sản phẩm, qua đó bảo vệ sức khỏe người tiêu dùng
21 CFR Part 11 được FDA (Cục Quản lý Thực phẩm và Dược phẩm Hoa Kỳ) thiết kế để quy định về việc sử dụng hồ sơ điện tử và chữ ký điện tử. Phần này đặt ra yêu cầu pháp lý cho các hồ sơ điện tử và chữ ký điện tử để chúng có thể được coi là tương đương với hồ sơ giấy và chữ ký viết tay về mặt pháp lý. Điều này áp dụng cho tất cả các công ty trong ngành công nghiệp y tế mà FDA có thẩm quyền quản lý, và nhằm đảm bảo tính toàn vẹn, độ tin cậy và độ chính xác của dữ liệu điện tử.
Cụ thể hơn, 21 CFR Part 11 hướng dẫn các nhà sản xuất dược phẩm về cách tạo, quản lý và duy trì hồ sơ điện tử và chữ ký điện tử. Tiêu chuẩn cũng đề cập về điều kiện mà các nhà sản xuất dược phẩm trở nên đủ điều kiện để tuân thủ quy định này.
Các nhà sản xuất thuốc cho các thử nghiệm lâm sàng, nghiên cứu tương dương sinh học và phân phối, phòng thí nghiệm, sản xuất theo hợp đồng, nhà cung cấp… phải tham khảo các hướng dẫn thực tế liên quan đến dữ liệu toàn vẹn để đảm bảo việc tuân thủ trong quản lý dữ liệu.
21 CFR Part 11 là một điều lệ có hiệu lực pháp lý mà các công ty phải tuân thủ khi tạo ra hồ sơ điện tử.
.jpg)
21 CFR Part 11
21 CFR Part 11 áp dụng cho các bản ghi điện tử được tạo, sửa đổi, duy trì, lưu trữ, truy xuất hoặc truyền tải theo yêu cầu được quy định bởi cơ quan
Các hồ sơ/chữ ký điện tử đáp ứng được yêu cầu của 21 CFR Part 11 có thể được sử dụng thay cho hồ sơ giấy
21 CFR Part 11 đại diện cho tiêu chuẩn được sử dụng nhiều nhất và cần thiết để quản lý dữ liệu một cách chính xác.
Tiêu chuẩn 21 CFR Part 11 của FDA bao gồm các quy định sau:
-
Quy định về Hệ thống điện tử: Các hệ thống sử dụng hồ sơ điện tử phải được kiểm soát, bao gồm việc kiểm tra, xác thực, bảo vệ và duy trì để đảm bảo tính toàn vẹn của hồ sơ.
-
Quản lý người dùng: Yêu cầu xác định và xác thực người dùng để đảm bảo rằng chỉ những người được ủy quyền mới có thể truy cập vào hệ thống.
-
Chữ ký điện tử: Cần phải có một phương pháp định danh duy nhất cho mỗi người sử dụng, như tên đăng nhập và mật khẩu, và yêu cầu chữ ký điện tử phải bảo đảm tính toàn vẹn của hồ sơ liên quan.
-
Bảo mật dữ liệu: Phải có các biện pháp bảo vệ để ngăn chặn việc truy cập trái phép hoặc thay đổi hồ sơ điện tử.
-
Lưu trữ dữ liệu: Hồ sơ điện tử phải được lưu trữ ở dạng có thể truy cập lại và tái tạo chính xác nội dung khi cần thiết.
-
Kiểm kê và theo dõi: Hệ thống phải có khả năng ghi lại và duy trì kiểm kê các sự kiện liên quan đến hồ sơ điện tử.
-
Đào tạo người dùng: Các tổ chức phải đào tạo người dùng của hệ thống để đảm bảo họ hiểu cách sử dụng hệ thống theo quy định.
-
Sao lưu và khôi phục: Các hệ thống cần có khả năng sao lưu và khôi phục hồ sơ điện tử.
-
Kiểm soát phiên bản: Cần phải có các biện pháp để quản lý các phiên bản của hồ sơ điện tử.
-
Audit trail: phải được thiết lập để ghi lại thời gian, người dùng và các thay đổi được thực hiện đối với hồ sơ điện tử.
.jpg)
Những quy định này giúp đảm bảo rằng hồ sơ điện tử và chữ ký điện tử được quản lý một cách an toàn, đáng tin cậy và có thể được kiểm tra một cách chặt chẽ, đồng thời duy trì sự tuân thủ đối với các yêu cầu quy định của FDA.
Cụ thể hơn các yêu cầu chính:
- ** Các quy định chung: bao gồm phạm vi của 21 CFR Part 11, cách triển khai nó và các định nghĩa liên quan. Điều này cũng xác định các điều kiện mà các nhà sản xuất dược phẩm trở nên đủ điều kiện để tuân thủ quy định này.
- ** Hồ sơ điện tử: 21 CFR Part 11 áp dụng cho các hồ sơ dưới dạng điện tử được tạo, sửa đổi, duy trì, lưu trữ, truy xuất hoặc truyền tải, theo bất kỳ yêu cầu hồ sơ nào được đặt ra trong các quy định của cơ quan.
- ** Chữ ký điện tử: các chữ ký điện tử và các hồ sơ điện tử liên quan phải đáp ứng các yêu cầu của phần này, cơ quan sẽ coi các chữ ký điện tử là tương đương với chữ ký viết tay đầy đủ, chữ viết tắt và các chữ ký chung khác được yêu cầu bởi các quy định của cơ quan, trừ khi được loại trừ cụ thể bởi quy định (các) có hiệu lực vào hoặc sau ngày 20 tháng 8 năm 1997.
---------
CÁC QUY ĐỊNH CHUNG VỀ THUẬT NGỮ
Thuật ngữ sau đây cung cấp cái nhìn tổng quan được sử dụng xuyên suốt tài liệu và các loại hồ sơ áp dụng và không áp dụng. Dưới đây là các thuật ngữ cần thiết để làm quen:
Bản ghi điện tử bao gồm văn bản, đồ họa, dữ liệu, âm thanh, hình ảnh, hoặc thông tin khác dưới dạng số được tạo ra, sửa đổi, duy trì, lưu trữ, truy xuất, hoặc phân phối bởi một hệ thống máy tính. Những bản ghi này phải chứa dữ liệu và metadata ở định dạng có thể đọc được. Ngoài ra, chúng phải sẵn sàng để truy xuất trong suốt toàn bộ thời gian lưu trữ.
I-Bản ghi điện tử ( Electronic records)
Bản ghi điện tử bao gồm văn bản, đồ họa, dữ liệu, âm thanh, hình ảnh, hoặc thông tin khác dưới dạng số được tạo ra, sửa đổi, duy trì, lưu trữ, truy xuất, hoặc phân phối bởi một hệ thống máy tính. Những bản ghi này phải chứa dữ liệu và metadata ở định dạng có thể đọc được. Ngoài ra, chúng phải sẵn sàng để truy xuất trong suốt toàn bộ thời gian lưu trữ.
Bản ghi điện tử đã ký phải có:
-
1- Tên in của người ký
-
2- Ngày và giờ khi chữ ký được thực hiện
-
3- Ý nghĩa của chữ ký (như xem xét, phê duyệt, trách nhiệm, hoặc tác giả)
II- Chữ ký số ( Digital signature)
** Chữ ký số ( chữ ký điện tử ) được định nghĩa là một chữ ký điện tử dựa trên các phương pháp mã hoá xác minh người tạo ra chữ kí, được tính toán bằng cách sử dụng một nhóm quy tắc và tham số sao cho có thể xác minh được người ký và tính toàn vẹn của dữ liệu. Chữ ký số phải đảm bảo rằng các hồ sơ điện tử có thể được xác thực và duy trì tính toàn vẹn, giúp chúng có giá trị pháp lý tương đương với chữ ký viết tay trên hồ sơ giấy.
** Các quy định 21 CFR Part 11 này yêu cầu đảm bảo chữ ký điện tử phải:
-
Được xác thực: Có các biện pháp để xác thực danh tính của người ký.
-
Độc nhất: Chỉ có thể sử dụng bởi người đã được ủy quyền.
-
Liên kết chặt chẽ với Hồ sơ điện tử: Không thể tách rời hoặc thay đổi mà không được phát hiện.
-
Bảo mật: Phải bảo vệ để ngăn chặn việc giả mạo hoặc sử dụng trái phép.
III-Audit trail (Dữ liệu gốc)
Là một hồ sơ được tài liệu hóa, theo thứ tự thời gian của các hoạt động hệ thống với chi tiết về cách thức các hoạt động này ảnh hưởng đến một hoạt động, quy trình, hoặc sự kiện cụ thể. audit trail bao gồm các hồ sơ được bảo mật, tạo ra bởi máy tính và có dấu thời gian, và chứa thông tin về ai, cái gì, khi nào và tại sao của hồ sơ. được tạo ra bằng máy tính và đóng dấu thời gian.
Nó phải ghi nhận thời gian người vận hành truy cập và hoạt động, sửa đổi hoặc xóa một bản ghi điện tử. cần phải có nhật ký theo dõi thay đổi hoạt động đầy đủ để theo dõi việc sử dụng chữ ký điện tử, đảm bảo tính an toàn theo thời gian hoặc một tập bản ghi, cung cấp bằng chứng dạng tài liệu về chuỗi các hoạt động có ảnh hưởng đến vận hành, quy trình hoặc sự kiện cụ thể tại bất kì thời điểm nào. FDA khuyến cáo các audit trail (dữ liệu gốc) ghi nhận các sự thay đổi đối với dữ liệu quan trọng phải được xem xét từng bản bản một trước khi phê duyệt lần cuối bản ghi.
IV -ALCOA
là một từ viết tắt được sử dụng bởi FDA, đại diện cho các thuật ngữ Attributable (Có thể quy cho), Legible (Có thể đọc được), Contemporaneous (Đồng thời), Original (Gốc), và Accurate (Chính xác). Khái niệm đằng sau ALCOA là chất lượng dữ liệu ảnh hưởng trực tiếp đến chất lượng sản phẩm, với trọng tâm đặt vào việc thực hiện các công việc một cách chính xác ngay từ lần đầu tiên và báo cáo kết quả ngay lập tức. Vì ALCOA được sử dụng trong nhiều tài liệu quy định của FDA, nên việc hiểu rõ ý nghĩa của mỗi thuật ngữ là quan trọng.
CÓ THỂ QUY CHO CÓ THỂ ĐỌC ĐƯỢC ĐỒNG THỜI GỐC CHÍNH XÁC
- Ai thu thập dữ liệu và khi nào hành động được thực hiện?
- Dữ liệu có thể đọc được bởi người khác trong suốt thời gian lưu trữ không?
- Dữ liệu được ghi chép tại thời điểm thực hiện hoạt động?
- Dữ liệu gốc hoặc dữ liệu nguồn có sẵn ở dạng gốc hoặc có bản sao chính xác không?
- Dữ liệu có chứa ngữ cảnh/ý nghĩa (tức là metadata) không?
Metadata là giải thích về dữ liệu mà nó đề cập đến. Ví dụ, bạn được cho một giá trị dữ liệu là "300". Metadata là thứ cung cấp ngữ cảnh cho giá trị này, tức là đó là số lượng hạt trên mỗi mét khối cho một máy đếm hạt.
V- EU GMP Phụ lục 11
Là một phần của Liên minh Châu Âu (EU), EudraLex là bộ sưu tập các quy tắc và quy định điều chỉnh các sản phẩm dược phẩm (cho cả sử dụng ở người và thú y). Phụ lục 11 là một phần của Hướng dẫn GMP Châu Âu và chứa các điều khoản tham chiếu cho các hệ thống máy tính được sử dụng bởi các tổ chức trong ngành công nghiệp dược phẩm. Lưu ý rằng Phụ lục 11 là một hướng dẫn, không phải là một quy định (21 CFR Phần 11 là một quy định).
Phụ lục 11 xác định các tiêu chí cho việc quản lý bản ghi điện tử và chữ ký. Những hướng dẫn này tương tự như của đối tác Hoa Kỳ. Sự xem xét trung tâm của cả EU GMP Phụ lục 11 và tài liệu 21 CFR Phần 11 là đảm bảo rằng các bản ghi được nhập một cách chính xác, không thể bị can thiệp, có thể được lưu trữ trong thời gian lưu trữ cũng như truy xuất (đầy đủ) bất cứ lúc nào trong quá trình sử dụng và trong thời gian lưu trữ. Trong mỗi quy định, có một trọng tâm mạnh mẽ vào độ chính xác, tính toàn vẹn, bảo mật và khả năng truy xuất của bản ghi.
Từ hướng dẫn, có một số thành phần quan trọng của một hệ thống quản lý dữ liệu phù hợp, tạo nền tảng cho các quy trình vận hành tiêu chuẩn (SOPs) sau đây:
SOP BẢO TRÌ HỆ THỐNG Bảo trì phù hợp phải được thực hiện theo cách kiểm soát.
SOP BẢO MẬT VẬT LÝ Phải có các biện pháp kiểm soát đảm bảo truy cập an toàn và ngăn chặn xâm nhập.
SOP BẢO MẬT LOGIC Phải có chính sách người dùng và mật khẩu.
SOP QUẢN LÝ SỰ CỐ VÀ VẤN ĐỀ Phải có cách thức để quản lý và giao tiếp về một vấn đề có thể xảy ra.
SOP KIỂM SOÁT THAY ĐỔI HỆ THỐNG Phải hiểu cách thức bất kỳ thay đổi nào có thể ảnh hưởng đến quy trình.
SOP PHỤC HỒI THẢM HỌA Phải có cách thức để đảm bảo dữ liệu được bảo vệ và quy trình có thể phục hồi trong tình huống thảm họa.
SOP SAO LƯU VÀ PHỤC HỒI Sao lưu dữ liệu thường xuyên phải được kiểm soát.
----------------------
3- Kiểm soát nhận dạng - Tài khoản và mật khẩu
- Những người sử dụng có ý định tạo hồ sơ điện tử/hồ sơ kỹ thuật số có chữ ký phải được kiểm soát theo các quy tắc nhận dạng. Mỗi người dùng phải có tài khoản duy nhất để xác định rõ ràng người dùng khi họ truy cập vào hệ thống.
- Đảm bảo kiểm tra / thay đổi mật khẩu định kì. Mật khẩu phải đủ mạnh và bảo mật để ngăn chặn việc truy cập không được phép.
- Đặc quyền ID sẽ được cấu hình riêng lẻ hoặc theo nhóm cơ bản; người vận hành, giám sát, quản trị viên sẽ có mức độ truy cập khác nhau
-
Quản lý tài khoản: Phải có quy trình để cấp phát, quản lý và thu hồi tài khoản người dùng, cũng như thay đổi mật khẩu khi cần thiết.
-
Theo dõi và kiểm soát: Phải theo dõi và kiểm soát việc truy cập của người dùng, bao gồm cả việc kiểm soát sau khi có s
- Cam kết chất lượng
- Bảo hành chính hãng
- Giao hàng tận nơi
- DỊCH VỤ 24/7
Có thể bạn thích
-
 Liên hệThêm vào giỏ hàng
Liên hệThêm vào giỏ hàng -
Hot
_-02-01-2024-10-19-51.jpg "Câu Hỏi Thường Gặp - Diễn đàn Thiết kế và Kỹ thuật Phòng Sạch Hàng năm lần thứ 3") Liên hệThêm vào giỏ hàng
Liên hệThêm vào giỏ hàng -
Hot
 Liên hệThêm vào giỏ hàng
Liên hệThêm vào giỏ hàng -
 Liên hệThêm vào giỏ hàng
Liên hệThêm vào giỏ hàng -
 Liên hệThêm vào giỏ hàng
Liên hệThêm vào giỏ hàng -
 Liên hệThêm vào giỏ hàng
Liên hệThêm vào giỏ hàng -
Hot
Liên hệThêm vào giỏ hàng
-
 Liên hệThêm vào giỏ hàng
Liên hệThêm vào giỏ hàng -
 Liên hệThêm vào giỏ hàng
Liên hệThêm vào giỏ hàng -
 là gì? Tại sao nó quan trọng với Xác minh và Toàn Vẹn Dữ Liệu?") Liên hệThêm vào giỏ hàng
Liên hệThêm vào giỏ hàng -
Hot
 đáp ứng các tiêu chuẩn của GMP & FDA") Giải pháp PMS tuân thủ Sự toàn vẹn của dữ liệu (Data Integrity) đáp ứng các tiêu chuẩn của GMP & FDALiên hệThêm vào giỏ hàng
Giải pháp PMS tuân thủ Sự toàn vẹn của dữ liệu (Data Integrity) đáp ứng các tiêu chuẩn của GMP & FDALiên hệThêm vào giỏ hàng -
_-27-12-2023-12-39-56.jpg "Tổng quan các cuộc thanh tra GMP EU, PIC/S và các nước SRA khác") Liên hệThêm vào giỏ hàng
Liên hệThêm vào giỏ hàng
.png)




